BIOSENSORS, MOTOR PROTEINS AND MORE
MARTIN WEBB
SCIENCE
Home > Other research
OTHER RESEARCH
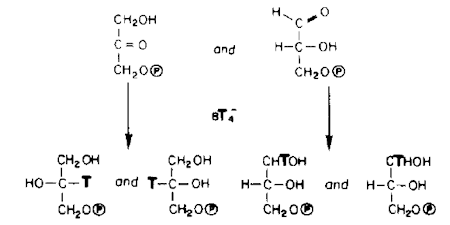
TRIOSEPHOSPHATE ISOMERASE
For my DPhil project, I worked in Jeremy Knowles' group (Oxford, then Harvard), using chemical biology techniques to study how substrates interacted with this "perfect" enzyme. Several projects within the Knowles Group were aimed at elucidating the kinetic mechanism and determining rate constants of elementary steps. This showed that the chemistry had rates similar to diffusion controlled binding of substrates.
High resolution structural information was not yet available. So my project used indirect methods to try to understand how the substrates bound and interacted with the amino acids to produce the very high catalytic rates.
Specifically, I looked at how the bound substrates (and substrate analogue) were accessible to external chemical reaction: this was by reduction of the bound substrate carbonyl by borohydride. How was the substrate reactivity modulated by interaction with amino acids? A major part of the project was developing methods to determine the stereochemistry of the product of this reduction. We also used 31P NMR to examine the bound substrates (Ref: 1, 2, 4)
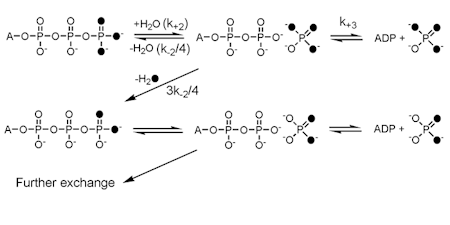
OXYGEN EXCHANGE
We use stable oxygen isotopes to follow the fate of particular oxygen atoms during ATPase and GTPase reactions. Typically, this uses [18O3]ATP and follows the exchange of oxygens with solvent water. Analysis is generally by mass spectroscopy, although 31P NMR at high resolution has also been used.
This gives information about the kinetics of various steps during these reactions, particulatly the reversibility of the on-protein cleavage step, that is bound ATP to bound ADP.Pi, and subsequent phosphate release.
Positional isotope exchange (PIX): an alternative isotope exchange protocol using oxygen-18 in the bridging oxygen between the beta- and gamma-Ps. This tests at which stage the cleavage reaction occurs during the overall ATPase reaction mechanism, since this bridging oxygen is only released when the gamma-phosphate is cleaved from ADP.
Applications of these methods have mainly been within the myosin project, but also on small G proteins and helicases
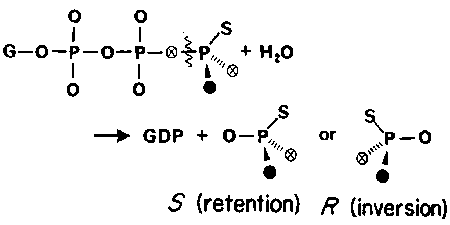
STEREOCHEMISTRY OF PHOSPHOTRANSFER
A general issue of the chemical mechanism of ATPases is whether there is a phosphoenzyme intermediate as opposed to direct transfer of phosphate. Such a phosphoenzyme may be present only transiently and so not in sufficient amount to identify directly.
The stereochemical course of phosphotransfer reactions was shown to provide a method that could distinguish the number of phosphotransfer reactions in an enzyme-catalysed pathway: each transfer occurs with inversion at the transferred P. In order to develop this method for phosphate hydrolysis, a way was needed to distinguish and so identify the four oxygen positions of Pi and hence its chirality. Only three stable isotopes of oxygen are available, and so the fourth position was substituted with sulphur.
Methods were developed to label ATPgS with 17O and 18O stereospecifically and to use 31P NMR to distinguish the enantiomers of (16O, 17O,18O)thiophosphate (Ref: 6, 8, 15)
The method was subsequently used to show for several ATPases and GTPases whether or not they had even transient phosphoenzymes on their pathways. (Ref: 8, 10, 11, 14, 16, 19, 20, 34)
Of particular note, this analysis was done for the mitochondrial F1 ATPAse reaction, showing there was direct, in-line displacement of ADP by water. This effectively discounted mechanisms requiring a phosphoenzyme for the mitochondrial ATP synthase. (Ref: 10)
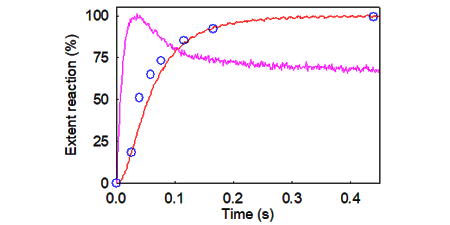
KINETIC METHODS
A major approach of my work has been to use rapid reaction techniques, to measure reactions in real-time and single turnover. These were particularly combined with flruorescence labeling to provide mainly fluorescence intensity signals, but also anisotropy.
Because the time scales of biological processes of interest are often on the millisecond to second timescale, this requires the use of rapid reaction techniques. We use two rapid mixing techniques, 'stopped flow fluorescence' and 'quenched flow'.
In stopped-flow, two reactants are mixed rapidly and a fluorescence signal is followed with time. There is also the potential of double mixing: two reactants are mixed and a reaction starts and continues until a third component is mixed.
In quenched flow measurements, the two reactants are mixed and the reaction is allowed to proceed for a particular time. The reaction is then quenched (usually in acid to denature proteins), for subsequent analysis of the extent of reaction by quantitative methods, such as hplc or gel electrophoresis.
Many of the publications use one or both of these methods.
We also used laser flash photolysis of caged compounds to initiate a reaction so that it can be followed on a millisecond timescale.